

摘要:采用基于密度泛函理论的平面波超软赝势方法对本征Zn2GeO4,Mn2+掺杂Zn2GeO4,Mn2+/N2-共掺杂Zn2GeO4超晶胞进行了几何结构优化,计算了掺杂前后体系的晶格常数、能带结构、态密度和光学性质.结果表明,Mn离子掺入后,Mn离子3d轨道与O离子2p轨道之间有强烈的轨道杂化效应,掺杂系统不稳定,而Mn/N离子共掺后,Mn离子和N离子之间的吸引作用克服了Mn离子之间的排斥作用,能够明显地提高掺杂浓度和体系的稳定性.光学性质计算结果表明,Mn离子与N离子共掺杂能改善Zn2GeO4电子在低能区的光学跃迁特性,增强电子在可见光区的光学跃迁;吸收谱计算结果显示,Mn离子与N离子掺入后体系对低频电磁波吸收增加.
 加入收藏
加入收藏
1、引言
锗酸锌(Zn2GeO4)是一种宽带隙半导体三元氧化物材料,具有良好的稳定性和光学性质,在场发射显示器、高性能紫外光电探测器和光催化剂中具有巨大的应用前景.近年来,人们对Zn2GeO4化合物光电性能的研究方兴未艾.然而,由于其带隙约4.68eV,本征Zn2GeO4晶体只对紫外光产生响应,这极大地限制了其实际应用前景.然而其宽带隙的特点也给人们提供了丰富的调制手段,在理想的可见光区,如何拓展其光吸收能力对提高Zn2GeO4半导体材料的应用具有重要的理论和实验研究意义[1,2,3,4].
掺杂离子是调节半导体电子结构和光学性质的有效手段之一.如果掺杂的离子合适,可以将Zn2GeO4晶体对紫外光响应扩展到可见光区域.锰离子作为稀土离子的替代物,许多研究人员通过实验的方法已经研究了Mn2+活化剂对Zn2GeO4晶体的光学调制行为,发现在Zn2GeO4中,由于Zn2GeO4的能量从Zn2GeO4宿主到四面体晶格点的Mn2+,产生了一种高效的绿色发光磷光体[5].
然而单一元素掺杂也会造成一些不足,如理想掺杂剂的溶解度有限,并在某些情况下自发形成补偿缺陷等.因此在这样的环境下,人们提出了共掺杂的理念,通过共掺杂来提高结构稳定性和光学性能,减少补偿缺陷来改变电子结构属性.如Zn2GeO4:Bi3+,Yb3+;Zn2GeO4:Mn2+,Yb3+;Zn2GeO4:Mn2+,Pr3+[6,7,8].对Zn2GeO4晶体而言,从我们掌握的文献资料中,目前还未发现有非金属离子掺杂的实验和理论研究.迄今为止,金属与非金属氮(N)共掺杂在TiO2半导体材料中得到了广泛的应用,如Ta/N-,Nb/N-,Br/N-,La/N共掺杂TiO2[9,10,11,12].其吸收边呈现出显著的红移,并对其在可见光下进行了良好的改性,制备出了具有良好的改性活性的光学材料.故在本文中,我们利用第一性原理,先从理论上揭示Mn离子掺杂对Zn2GeO4晶体电子结构和光学性质影响,分析N离子对Zn2GeO4光电性能的调制,再讨论Mn/N离子共掺杂对Zn2GeO4晶体电子结构和光学性能产生的影响.
2、模型和计算方法
基于第一性原理平面波赝势方法,利用局域密度近似(LDA)交换-关联能,理论模拟由CASTEP量子力学模块计算软件完成.参与计算的价态电子是:Zn3d104s2,Ge4s24p2,O2s22p4,Mn3d54s2,N2s22p3,平面波截止能量(Cutoff-energy)取360eV,k网格(k-mesh)的大小4×4×2.收敛精度为离子间最大相互作用力0.02eV/nm和离子平均能量5.0×10-6eV.对于单Mn、N掺杂的系统,在宿主晶胞内的42个离子的掺杂浓度为2.38%,化学计量学分别为Zn1.92Mn0.08GeO4和Zn2GeO3.83N0.17,在Mn/N共掺杂系统中,化学计量表达为Zn1.92Mn0.08GeO3.83N0.17.
3、结果和讨论
3.1几何结构
如图1所示,本征Zn2GeO4属于具有菱形晶胞的典型硅锌矿结构(a=0.8836nm,R=107.79.).这些离子沿着c轴以六个相等分隔的层排列成层,并且每个Ge和Zn离子处于四个氧离子的四面体环境中.优化结果表明,一种GeO4四面体和两种ZnO4四面体是通过边缘氧离子相互结合,多面体中不同的Ge-O和Zn-O键表明GeO4和ZnO4四面体结构是扭曲的,这与实验结果一致[13].
图1本征、Mn掺杂和Mn/N共掺杂Zn2GeO4晶体结构
表1显示Mn掺杂导致Zn2GeO4晶胞体积稍微增大,N掺杂Zn2GeO4结构中,引入的N离子键合到一个Ge和两个Zn离子上,晶格常数均增大且不相等;Mn/N离子共掺杂系统,相对本征Zn2GeO4晶体而言体积变化最为明显.体系结合能E反映出晶体在进行几何优化后结构的稳定性,总能量E越小,对应的晶体结构稳定性越好.表1表明,Mn离子掺杂导致系统不稳定,N离子的掺杂能够有效地提高Zn2GeO4结构稳定性,而Mn/N共掺杂Zn2GeO4晶体具有更好的稳定性.
表1Zn2GeO4晶体结构常数和结合能
通常情况下,由不同化学键构成的内部扭曲多面体会引起内部偶极矩和局部静电场,这可以促进由半导体光照产生的电子-空穴对的分离.为了揭示离子掺杂引起的Zn2GeO4晶胞结构的变化,我们分析了晶体中主要化学键的电子集居数分布情况.显然,本征Zn2GeO4体中ZnO4和GeO4四面体的键长是相似的,因此内部偶极矩很小.Mn掺杂的Zn2GeO4体系中,Mn-O键的电荷集居数(0.41e-0.45e)相较于Zn-O键的电荷集居数(0.23e-0.58e)而言没有明显影响,但是Mn离子掺杂导致Ge-O键的电荷集居数(0.25e-0.36e)远小于本征Zn2GeO4体中Ge-O键的电荷集居数(0.55e-0.61e),这意味着Mn离子的掺杂对ZnO4四面体的结构几乎没有影响,而对GeO4四面体的结构影响比较大,从而导致Zn2GeO4晶胞体积增加;N掺杂的Zn2GeO4体系中,N-Zn键的电荷集居数(0.64e和0.69e)稍大于O-Zn键的电荷集居数(0.38e-0.64e),表明N和Zn离子之间的相互作用比Zn-O之间的相互作用强.此外,电子集居数为0.35e的共价键N-Ge键也比Ge-O键强.因此,由于四面体中电荷的不均衡分布,N掺杂体系的偶极矩得到增强.在Mn/N共掺杂体系中,Zn-N键的电子集居数(0.85e和0.99e)明显大于Zn-O键的电荷集居数(0.42e-0.58e),Ge-N(0.49e)也强于Ge-O键的电荷集居数(0.12e-0.42e),因此可推测由于掺杂体系具有不平衡的化学键,从而导致基本结构单元中存在较大的电偶极矩,造成Mn和N共掺杂Zn2GeO4体积变化最为明显.然而这种内部静电场的出现和偶极矩的增强,有利于晶体内部电荷载体的分离和结构的稳定性.
表2本征和掺杂Zn2GeO4晶体电荷集居数分布(单位:e)
3.2电子结构
3.2.1本征Zn2GeO4电子结构分析
为分析和比较掺杂对Zn2GeO4电子结构的影响,先计算了本征Zn2GeO4的电子结构、态密度和差分电荷密度分布.图2(a)能带结构的计算结果表明Zn2GeO4的带隙较大,价带顶部要高于费米能级,属于直接带隙的P型半导体类型.导带底的态密度变化较小,说明导带底的电子有效质量较小;价态顶态密度变化较慢,价态中空穴有效质量大.Zn2GeO4的价态由-7.7eV—-7.5eV的下价态和-6.2eV—0.3eV的上价态组成,在价态顶有兼并的重空穴和自旋-轨道耦合所分裂出的劈裂带.由图2(b)的分波态密度可知,价态顶主要有O离子的2p态组成,导带主要是Zn和Ge的4s态组成.图2(c)中的差分电荷密度分布表明,Ge和O离子之间的相互作用明显的强于Zn与O离子,Zn-O键表现出明显的离子键特征,而Ge-O键之间体现出共价键的特性.表2中电子集居数分布也证明了这样的特点.
3.2.2Mn掺杂Zn2GeO4电子结构分析
图3为优化后的Mn掺杂Zn2GeO4晶体结构的能带结构及分波态密度.从图3(a)可知,掺杂Mn后Zn2GeO4晶体的导带底下移到费米能级以下,仍然是直接带隙半导体.对比图3(b)分波态
图2Zn2GeO4晶体态密度(a)、分波态密度(b)和差分电荷密度分布(c)
密度可知,Mn掺杂Zn2GeO4的下价带主要由O离子的2s态组成,Ge离子的4s、4p态也有所贡献,上价态主要由Zn离子的4s态、Ge离子的4s态和O离子的2p态杂化而成.主要变化来自导带,导带主要由Zn和Ge的4s态与Mn离子的3d态组成,导带底下移主要来源于Mn离子的3d态的贡献.对比图3(b)和图2(b)可知,Mn掺入后对Zn和Ge的态密度峰值影响不大,但是它们的态密度变弥散,向低能方向移动;同时导带底越过费米能级形成简并态,在导带底附近存在多余的电子,说明Mn掺杂可获得n型Zn2GeO4晶体材料.但杂质能级中电子间的互相作用导致其局域在导带底附近,费米能级附近电子增加,离子间排斥作用增强,从而导致Zn2GeO4晶体的体积增大.图3(c)中电子密度差分分布直观地表明了Mn离子掺杂Zn2GeO4中引起了各个离子周围电子环境的变化,Mn2+离子的引入促进各离子间形成更多的共有化区域,特别是造成了Mn离子和Ge离子间轨道重叠,这有利于更多的电子脱离离子束缚,使得更多的电子成为共有化电子,进而使其带隙变窄,实现了对Zn2GeO4晶体带隙的调制.
图3Mn掺杂Zn2GeO4晶体态密度(a)、分波态密度(b)和差分电荷密度分布(c)
3.2.3N掺杂Zn2GeO4电子结构分析
氮元素是常用来实现p型掺杂的杂质,为此我们在优化后计算了N掺杂Zn2GeO4的电子结构.图4(a)表明N掺杂使得Zn2GeO4的导带向低能端有较明显的移动,这导致了Zn2GeO4晶体的能带间隙减小,同时这也有利于电子和空穴由价带顶向导带底的跃迁.图4(b)揭示了N掺杂Zn2GeO4最主要的影响来源于价带顶,费米能级附近形成了新的局域态密度峰,该态密度峰来自掺杂N离子的2p态,它排斥Zn2GeO4上的价态电子,使其向高能端移动.N离子2p态打破了Zn2GeO4晶体价带顶的p-d轨道杂化,而且N离子2p态电子比O离子的2p态少一个电子,因此掺杂N可以在Zn2GeO4的价带顶形成更多的空穴,从而提高Zn2GeO4晶体的p型导电能力.图4(c)显示N离子掺杂Zn2GeO4对各离子周围环境影响不大,这主要因为N离子和O离子的离子半径和非金属性差别不大.
3.2.3Mn和N共掺杂Zn2GeO4电子结构分析
图5为Mn和N共掺杂Zn2GeO4的能带结构、态密度分布和电子差分密度图.图5(a)揭示Mn和N离子共掺杂后,Zn2GeO4导带分布表现出了较高的弥散性,而这种弥散性分布主要来源于Mn3d态的电子;价带顶分布于费米能级之上,这主要来源于N离子的2p态的贡献.图5(b)表明,Zn离子态密度峰值略有升高,价带和导带均向低能级方向移动;相对于本征Zn2GeO4晶体,Ge离子态密度峰值有所降低,价带和导带几乎没有移动;Mn离子的3d态劈裂成两个独立的峰,其中一个峰进入费米能级;N离子的2p态位于费米能级之上;Mn与N共掺杂Zn2GeO4晶体费米能级进入价带顶,使其价带顶附近存在多余的空穴,与价带顶形成杂质能级,且杂质能级具有较强的局域态,这主要由N和O离子的2p态与Mn离子3d态轨道杂化形成.
图5Mn、N共掺杂Zn2GeO4晶体态密度(a)、分波态密度(b)和差分电荷密度分布(c)
图5(c)差分电荷密度分布表明,由于O离子电负性比N离子强,Mn离子3d态电子偏向与N离子成键,进而Zn—O键增强、Mn—O键减弱,O离子价电荷分布偏向Zn离子而偏离Mn离子,因此Mn离子与O间离子轨道交叠区域和费米能级处态密度重叠减小,降低Mn离子的受主特性,在费米能级附近,Mn离子3d轨道与N离子2p轨道杂化被增强.N离子不是p型材料的有效复合中心,主要起到对Mn离子的激活作用.共掺杂后Mn离子与N离子的吸引作用可克服Mn离子间的互相排斥,能有效提高受主掺杂浓度与系统的稳定性,Mn离子与N离子共掺杂可实现p型Zn2GeO4的高浓度掺杂.
4、Mn/N共掺杂Zn2GeO4的光学性质分析[14,15,16,17,18]14-18]
在线性响应范围内,介电函数ε(ω)可以用来表征固体宏观光学响应特性.通过对介电常数ε(ω)特点的分析,可以掌握电磁辐射在介质中的传播规律,了解其能带结构和其它光谱性质之间的联系.介电常数可表示为ε(ω)=ε1(ω)+iε2(ω),其中
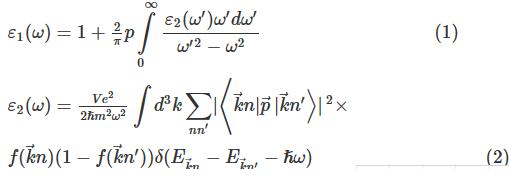
图6Zn2GeO4晶体介电常数(a)和光学吸收谱(b)下载原图
其它光学常数求解如下:
吸收系数: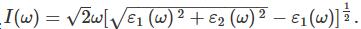 式中ε1(ω)、ε2(ω)分别为介电函数实部和虚部,m为自由电子质量,e为电子电量,ω为入射光子频率.
式中ε1(ω)、ε2(ω)分别为介电函数实部和虚部,m为自由电子质量,e为电子电量,ω为入射光子频率.
图6(a)为Mn与N共掺杂前后Zn2GeO4的介电函数虚部ε2,ε2是联系带间跃迁微观物理过程与固体电子结构的桥梁,是电子在能级间跃迁的体现,ε2曲线介电峰的来源可通过能带结构与态密度解释.从图6(a)可以看出,本征Zn2GeO4的介电函数阈能约为4.2eV,这是基本吸收边,当光子能量高于4.2eV时ε2的值迅速上升.Zn2GeO4的介电函数虚部有两个特征峰,峰1与峰2分别对应能量为9.6eV与14.7eV.峰1对应直接跃迁阈,是O2p电子向Zn3d态跃迁;峰2是Zn2GeO4态密度图中上价带相对较弱的峰代表的能态与导带能态间电子跃迁所致.掺入Mn离子后,在2.6eV增加介电峰3,之前的两个介电峰峰值增强,峰3是价带电子向杂质带跃迁引起的,主要是O2p态与Mn3d态跃迁的结果,这表明Mn离子掺杂引起能级间跃迁电子数目上升;掺入N离子后峰值降低,这表明N离子掺杂对Zn2GeO4的电学性质有一定的抑制作用.在低能区,Mn/N共掺杂Zn2GeO4的光跃迁强度大于未掺杂Zn2GeO4,表明Mn/N共掺杂可改善Zn2GeO4低能区的光学跃迁,增强电子在可见光区的光学跃迁.
图6(b)是掺杂前后Zn2GeO4的吸收谱,结果表明,本征Zn2GeO4晶体在可见光区无光吸收,因其禁带宽度大于可见光的光子能量.Mn/N掺入后,因为禁带中引入了杂质能级,增强了晶体在可见光区的光吸收.本征Zn2GeO4吸收主峰的能量为9.6eV,Mn/N掺入后于低能区出现一新吸收峰,与介电函数虚部在低能区的变化相对应,与本征介电函数虚部相比,Mn/N共掺后吸收峰增强.
5、结论
基于密度泛函理论对本征Zn2GeO4,Mn离子单掺杂以及Mn/N共掺杂Zn2GeO4电子结构和光学性质进行了计算与分析.结果表明:掺杂后晶格体积增大,系统能量降低;Mn离子单掺杂及Mn/N离子共掺杂后Zn2GeO4仍属于直接带隙半导体;Mn离子掺入后Mn离子3d电子与邻近的O离子2p电子之间有强烈的轨道杂化效应,掺杂系统不稳定,较难实现理想的n型Zn2GeO4晶体;Mn/N离子共掺杂后,Mn离子与N离子的吸引作用克服了Mn离子间的排斥作用,可提高掺杂浓度和系统稳定性,Mn离子与N离子共掺杂可实现p型Zn2GeO4的高浓度掺杂.光学性质分析中,Mn离子与N离子共掺杂能够改善Zn2GeO4电子在低能区的光学跃迁特性,增强电子在可见光区的光学跃迁,对低频电磁波吸收增加.
参考文献:
[1]赵淑婷,李文琪,牟博石,等.Zn2GeO4:Mn2+绿色磷光粉的制备和长余辉性能[J].发光学报,2019,40:189.
[2]杨扬,丛妍,朱子茂,等.Zn2GeO4纳米棒的制备及发光性质[J].无机化学学报,2017,33:1757.
[12]张晓旭,马梅,张文蕾,等.La-N共掺杂锐钛矿相TiO2的电子结构及光学性质的第一性原理研究[J].原子与分子物理学报,2016,33:1113.
[13]吴成国,武文远,龚艳春,等.高压下Zn2GeO4带隙变化的第一性原理研究[J].物理学报,2015,64:114213.
[14]代武春,伏春平,孙凌涛,等.Mn-N共掺杂TiO2电子结构和光学性质的第一性原理计算[J].原子与分子物理学报,2016,33:345.
蔡宏宇,吴海涛,吴成国,何苏红.Mn~(2+)/N~(2-)共掺杂Zn_2GeO_4晶体电子结构和光学性质的研究[J].原子与分子物理学报,2020,37(05):749-755.
基金:陆军工程大学基础学科培育基金重点项目.
分享:


多孔材料广泛应用于过滤、分离、纯化、萃取、冷却、干燥和催化等领域,包括材料科学、工程、力学、地球科学和生物学等。沸石、黏土、陶瓷和木炭等许多常见的物质都是重要的多孔材料,其主要特征就是相互连通和永久性的孔道。沸石是在无机化学领域应用非常广泛的材料之一,作为一种具有很大商业价值的材料,它在气体分离和工业催化等领域有非常重要的应用。
2024-11-13
无机化学课程在高校化学的课堂教学中第一门公共基础课程,对于理工科学科,尤其是食品酿造学科的本科应用技术型大学,无机化学实验课程是一门必修基础课程[1]。对于公共基础课程存在着学生层次不同,内容设置单一和、专业特色不明显以及评价体系难以量化,不能充分发挥实验课程优势,难以激发学生积极性和主动性,不能很好引导学生将理论知识与实践充分结合[2]。
2020-09-15
本文将“雨课堂”与《无机及分析化学》课程相融合,通过“雨课堂”这个有效的教学平台实现了优秀教育资源的共享,使教师与学生展开了有效的课前、课中和课后互动,提升课程的教学效果,使学生能够扎实的掌握《无机及分析化学》的基础知识,为后续课程的学习提高有力的保障。
2020-09-09
基于《无机及分析化学》理论课程开设的《无机及分析化学实验》旨在加深学生对无机及分析化学基本理论的理解,更系统地学习、理解无机化学与分析化学实验的基本知识、基本操作和基本技能,养成严谨的、实事求是的科学态度,初步掌握实验研究的方法,为学习后续课程和将来从事实际工作打下良好的基础[1,2]。
2020-09-09
半导体光催化技术因其仅利用太阳能作为能源来降解污染物而被认为是一种很有前途的解决环境污染问题的技术。尽管人们对TiO2、ZnO和WO3等半导体做了大量研究,但这些半导体的光催化效率仍低于商业化所需的效率。限制效率的因素包括:(1)对太阳光不能完全吸收利用;(2)催化剂的稳定性差;(3)快速的电子-空穴复合速率;(4)缓慢的表面氧化还原反应速率。
2020-09-05
在过去的30年中,锂离子电池已广泛应用于便携式电子设备当中,并在近年来扩展到大型储能电站和电动汽车等应用领域。但是,锂离子电池在这些新的应用场景中需要在能量密度、安全性和使用寿命上满足更高的要求。石墨类碳材料作为最常见的商业化的负极材料,其较低的嵌锂电位会导致电池负极表面析锂现象的产生,从而给电池带来安全上的隐患;同时,石墨材料较低的比容量也限制了电池能量密度的进一步提高。
2020-09-05
稀土配位聚合物的合成与性质研究是当前稀土配位化学研究的热点领域之一,这类配合物在光致发光、电致发光、荧光探针、磁性、吸附、催化等材料领域具有潜在的应用价值。铕和铽在稀土配合物发光材料方面是应用最多的2种元素,一方面这2种元素三价离子的发光处于可见光区,分别能发射三基色中的红色和绿色光;另一方面,使用它们作为配位中心离子时,理论上可得到内量子发光效率非常高的发光材料。
2020-09-05
金属配合物由于其独特组装结构,在催化剂、传感器、气体吸附、发光材料、抗癌药物和抗菌等方面都有潜在的应用价值,因此一直以来都受到广泛的关注。含有吡啶基团的酰腙Schiff碱化合物,由于吡啶环上的氮原子的亲核能力很强,酰腙Schiff碱化合物易与不同的过渡金属离子以不同的配位模式作用,组装成结构新颖的酰腙Schiff碱配合物。
2020-09-05
分子基发光材料因其易于分子裁剪、结构丰富、颜色可调、性能可控、制备简单、能耗低等优点,近年来已成为新材料研究领域中的热点之一,在光电、信息、环境乃至生命科学等诸多高新技术领域具有广泛应用。在分子基发光材料的开发和设计研究中,荧光量子产率的提升、发射光谱和荧光寿命的调控等是该领域的关注重点之一。在众多分子基发光材料中,氟硼配合物因其优异的发光性能备受关注。
2020-09-05
针对粉体MOF衍生材料存在制备工艺复杂、薄膜厚度难以控制等问题,我们通过液相外延分步生长法制备了一种金属有机框架薄膜(PIZA-1),然后以其作牺牲模板,在惰性氛围中制备了一种CoSe2和N共掺杂的碳膜(CoSe2/N-CF),并用作DSSC对电极,其具有制备简单、粘结力强、厚度可调等优势。系统表征了CoSe2/N-CF形貌特点、结构性质及电化学性能,并深入研究了不同厚度薄膜、CoSe2颗粒大小对DSSC的光伏性能的影响。
2020-09-05
人气:5064

人气:4853

人气:3889

人气:3686

人气:2607
我要评论
期刊名称:化学学报
期刊人气:5671
主管单位:中国科学院
主办单位:中国科学院上海有机化学研究所,中国化学会
出版地方:上海
专业分类:化学
国际刊号:0567-7351
国内刊号:31-1320/O6
邮发代号:4-209
创刊时间:1933年
发行周期:月刊
期刊开本:大16开
见刊时间:一年半以上


影响因子:2.741

影响因子:1.160

影响因子:1.215
影响因子:0.770
影响因子:0.374
科普杂志阅读、期刊信息查询、论文下载、文献查询、原创文章检测等多项服务。
服务热线
您的论文已提交,我们会尽快联系您,请耐心等待!
你的密码已发送到您的邮箱,请查看!

 2020-06-17
2020-06-17
 431
上传者:管理员
431
上传者:管理员





